
Adrenoleukodystrophy
| Prevalence | The prevalence of X-linked adrenoleukodystrophy (X-ALD) is 1 in 20,000 to 50,000 individuals worldwide. This condition occurs with a similar frequency in all populations[1]. |
| Natural History | 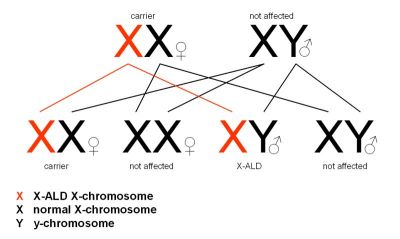 X-ALD is an X-linked recessive inherited condition that primarly affects males (females are carriers). It is due to mutations in ABCD1, a gene located on the X chromosome, which codes for adrenoleukodystrophy protein (ALDP), an integral peroxisomal membrane protein that transports VLCFacyl-CoA esters from the cytosol into the peroxisome[2,3]. Mutations in ABCD1 lead to abnormal accumulation of saturated unbranched very long chain fatty acids (VLCFAs)[4]. This accumulation is toxic in all tissues and causes oxidative stress and oxidative damage to proteins, microglial activation, and apoptosis[2]. The accumulation is greatest in nervous tissue white matter, adrenal cortex, testis, and in some lipid fractions (e.g. cholesterol esters of the adrenal gland and brain)[4]. X-ALD is an X-linked recessive inherited condition that primarly affects males (females are carriers). It is due to mutations in ABCD1, a gene located on the X chromosome, which codes for adrenoleukodystrophy protein (ALDP), an integral peroxisomal membrane protein that transports VLCFacyl-CoA esters from the cytosol into the peroxisome[2,3]. Mutations in ABCD1 lead to abnormal accumulation of saturated unbranched very long chain fatty acids (VLCFAs)[4]. This accumulation is toxic in all tissues and causes oxidative stress and oxidative damage to proteins, microglial activation, and apoptosis[2]. The accumulation is greatest in nervous tissue white matter, adrenal cortex, testis, and in some lipid fractions (e.g. cholesterol esters of the adrenal gland and brain)[4]. |
| Disease Presentation | X-ALD has varying phenotypes:
Approximately 50% of patients with X-ALD will develop the cerebral form of X-ALD at some point during their lifetime. The disease most frequently presents in childhood, typically between age 4 and 8 years, with the first noticable symptom being a decline in school performance. The early clinical features of cerebral ALD are often misdiagnosed as Attention Deficit Hyperactivity Disorder (ADHD). Onset of the disease involves deficits in cognitive ability, including:
Cerebral ALD progresses rapidly, and the neurological deficits quickly become more apparent. They include:
Within 2 to 3 years, individuals with cerebral ALD will have severe neurological disabilities that cause them to become bedridden, blind, unable to speak or respond, and require full-time nursing care and feeding by nasogastric tube or gastrostomy. Shortly after, these individuals die[2,3,5]. Adrenomyeloneuropathy (AMN) Virtually all patients with X-ALD who reach adulthood develop AMN, usually in the 3rd or 4th decade of life. Their symptoms are limited to the spinal cor and peripheral nerves, and include:
“Addison-Only” Phenotype Approximately 20% of male X-ALD patients have an “Addison-Only” phenotype which presents as adrenocortical insufficiency without clinical or MRI evidence of neurological involvement. This may be the presenting symptom in boys and men years or even decades before the onset of neurological symptoms[2,3,5]. Asymptomatic and Pre-Symptomatic Patients Individuals who are asymptomatic or pre-symptomatic are typically under age 7 years. They have normal cognitive function and are the key group for therapeutic interventions. Those who are pre-symptomatic are almost all at risk to develop neurologic (cerebral ALD or AMN) or endocrinologic (Addison’s disease) symptoms[2]. Female Carriers X-ALD is an X-linked recessive disorder, therefore females are carriers of the condition. Greater than 50% of female carriers exhibit some kind of abnormality on neurologic examination, most likely due to skewed X-inactivation in neuronal cells. Neurologic symptoms in carrier females have a typical onset between the 4th and 5th decade of life. Symptoms in female carriers are similar to those observed in males with AMN, and include:
|
| Effect on Fertility | In 1990, hematopoietic stem cell transplantation (HSCT) led to stabilization and possibly reversal of neurological changes in a boy with early childhood cerebral X-ALD. The mechanisms of benefit have not yet been identified as donor cells do not enter the central nervous system (CNS). However, gradual replacement of a portion of perivascular microglia by donor derived cells may exert favorable metabolic effect, or the favorable effect may be due to the immunosuppression and reconstitution of the immune system by donor derived cells. Regardless, HSCT is strongly recommended for patients at early stages of cerebral X-ALD, but not for those in more advanced stages. The 5 year survival rate for those in early stages of illness is 92%[5].
The goal of HSCT is to cure the patient from the primary disease and restore complete physical and mental health condition; however, infertility is common after myeloablative HSCT conditioned with total body irradiation (TBI) and high doses of gonadotoxic drugs. TBI and gonadotoxic drugs are typically used to condition patients prior to HSCT, both of which lead to absence of spermatozoa. In fact, TBI is the main cause of azoospermia in patients treated with HSCT. This is due to the fact that spermatogenic stem cells are more sensitive to chemotherapy and radiation than later stage germ cells, preventing the generation of new sperm cells[6]. Rovo et al. (2013) conducted a European Group for Blood and Marrow Transplantation (EBMT) large retrospective analysis of men who underwent HSCT and found that 81% of males receiving TBI presented with azoospermia and only 1% had normal sperm counts in follow-up sperm analysis. Anserini et al. (2002) found that azoospermia is less frequent in patients conditioned with busulphan and cyclophosphamide (50%) and uncommon in those treated with cyclophosphamide alone. Suggesting, perhaps, that busulphan and cyclophosphamiade or cyclophosphamide alone may affect fertility less than traditional HSCT conditioning with TBI and gonadotoxic drugs. |
| Fertility Preservation |  Given the risk of azoospermia is very high after HSCT, fertility preservation options should be discussed with all young men of reproductive age and parents of children facing HSCT[6].Boys with X-ALD are typically prepubertal at the time of HSCT and prepubertal males cannot produce semen for cryopreservation. Testicular tissue cryopreservation is currently the only option for prepubertal males at high risk for infertility interested in preserving future fertility[9]. Ginsberg et al. (2014) consented 57 families to testicular biopsy, and 28 children underwent the procedure with 1 experiencing a post-operative side effect. Parents who faced this fertility preservation option at their child’s diagnosis reported they made an informed decision and carefully weighed the risks and benefits of testicular tissue cryopreservation. Given the risk of azoospermia is very high after HSCT, fertility preservation options should be discussed with all young men of reproductive age and parents of children facing HSCT[6].Boys with X-ALD are typically prepubertal at the time of HSCT and prepubertal males cannot produce semen for cryopreservation. Testicular tissue cryopreservation is currently the only option for prepubertal males at high risk for infertility interested in preserving future fertility[9]. Ginsberg et al. (2014) consented 57 families to testicular biopsy, and 28 children underwent the procedure with 1 experiencing a post-operative side effect. Parents who faced this fertility preservation option at their child’s diagnosis reported they made an informed decision and carefully weighed the risks and benefits of testicular tissue cryopreservation.
Testicular Tissue Cryopreservation Alternatively, males with X-ALD who survive to adulthood may choose to use donor sperm or consider adoption. |
| Additional Considerations | Given that X-ALD is an inherited disorder, affected individuals and carriers may wish to discuss premplantation genetic diagnosis (PGD) and/or prenatal testing (chorionic villus sampling or amniocentesis) with a genetic counselor. |
| Patient Fact Sheet | Print the Oncofertility Consortium Fertility Preservation Patient Fact Sheet on Adrenoleukodystrophy (X-ALD) to provide to your patients and their families who are considering fertility preservation prior to HSCT treatment. This fact sheet is tailored specifically to the fertility needs of children with X-ALD.
Adrenoleukodystrophy Fertility Preservation Patient Fact Sheet |
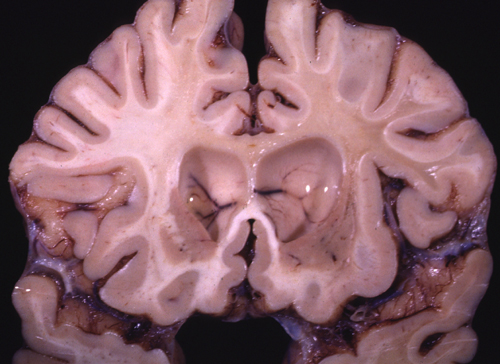 Cerebral Adrenoleukodystrophy
Cerebral Adrenoleukodystrophy 
References
[1] X-linked adrenoleukodystrophy. (2014). U.S. National Library of Medicine.http://ghr.nlm.nih.gov/condition/x-linked-adrenoleukodystrophy. Retrieved June 5, 2014.
[2] Engelen, M., Kemp, S., Visser, M. d., van Geel, B. M., Wanders, R. J., Aubourg, P., et al. (2012). X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet Journal of Rare Diseases, 7(12), 51.
[3] Kemp, S., & Wanders, R. (2010). Biochemical Aspects of X-Linked Adrenoleukodystrophy. Brain Pathology, 20, 831-837.
[4] Moser, H. W. (1997). Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain: a Journal Of Neurology, 120, 1485-1508.
[5] Moser, H. W. (2006). Therapy of X-linked adrenoleukodystrophy. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics, 3(2), 246–53. doi:10.1016/j.nurx.2006.01.004
[6] Tichelli, A., & Rovo, A. (2013). Fertility issues following hematopoietic stem cell transplantation. Expert Rev Hematol., 6(4), 375-388.
[7] Rovo, A., Aljurf, M., Chiodi, S., S, S., Salooja, N., & G, S. (2013). Ongoing graft-versus-host disease is a risk factor for azoospermia after allogeneic hematopoietic stem cell transplantation. A survey of the late effect working party of the European Group for Blood and Marrow Transplantation. Haematologica, 98(3), 339-345.
[8] Anserini, P., Chiodi, S., Spinelli, S., Costa, M., Conte, N., Copello, F., & Bacigalupo, A. (2002). Semen analysis following allogenic bone marrow transplantation. Additional data for evidence-based counselling. Bone Marrow Transplant, 30(7), 447-451.
[9] Ginsberg, J., Li, Y., Carlson, C., Gracia, C., Hobbie, W., Miller, V., . . . Kolon, T. (2014). Testicular tissue cryopreservation in prepubertal male children: An analysis of parental decision-making. Pediatr Blood Cancer.
About the Author
Allison Goetsch, MS, CGC is a pediatric genetic counselor at Ann and Robert H. Lurie Children’s Hospital of Chicago and a member of the Oncofertility Consortium administrative core team. She completed her graduate thesis with Dr. Teresa K. Woodruff researching oncofertility and hereditary breast and ovarian cancer (HBOC) syndrome. Allison’s primary goals are to increase fertility preservation awareness and education for both health care providers and patients regarding malignant and non-malignant diseases (and/or treatments) which threaten fertility.
This page was last updated February 6th, 2016.