
DiGeorge syndrome (22q11.2 deletion syndrome)
| Prevalence | DiGeorge Syndrome (22q11.2 deletion syndrome), which is also referred to as velocardiofacial syndrome, is one of the most common genetic syndromes with a prevalence of 1:4000 [1]. With approximately 2.5 million children born each year in the United States, it is estimated that around 500 to 750 new cases of DiGeorge Syndrome will be identified yearly [2]. |
| Natural History | 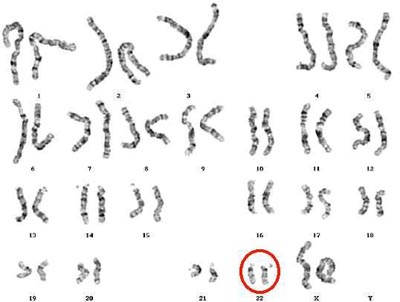 DiGeorge syndrome is caused by a 1.5-3 Mb hemizygous deletion of chromosome 22q11.2. Chromosome 22 has been found to possess a high number of low copy number repeats, which suggests responsibility for the instability of 22q11. In a majority of cases of DiGeorge Syndrome, the deletion is mediated by homologous recombination between these low copy number repeats [4] [5]. Within this deletion region is a key gene for early vertebrate development, TBX1. Haploinsufficiency of TBX1 is responsible for major phenotypes that are seen in individuals with DiGeorge syndrome. However, the variability of phenotypic features with a deletion of TBX1 suggests that altered interaction with downstream genes and environmental effects also affect the disease presentation. [6]. DiGeorge syndrome is caused by a 1.5-3 Mb hemizygous deletion of chromosome 22q11.2. Chromosome 22 has been found to possess a high number of low copy number repeats, which suggests responsibility for the instability of 22q11. In a majority of cases of DiGeorge Syndrome, the deletion is mediated by homologous recombination between these low copy number repeats [4] [5]. Within this deletion region is a key gene for early vertebrate development, TBX1. Haploinsufficiency of TBX1 is responsible for major phenotypes that are seen in individuals with DiGeorge syndrome. However, the variability of phenotypic features with a deletion of TBX1 suggests that altered interaction with downstream genes and environmental effects also affect the disease presentation. [6].
As in all microdeletion syndromes, inheritance is autosomal dominant, however most cases of DiGeorge syndrome result from a de novo microdeletion [1]. |
| Diagnosis | DiGeorge Syndrome is commonly diagnosed prenatally. Current indications for prenatal testing for 22q11.2 include a previous child with a 22q11.2 deletion; even though this recurrence risk may be low there is a possibility for germline mosaicism, an affected parent with a 22q11.2 deletion or a visible congenital heart defect seen on a prenatal ultrasound [7]. If a congenital heart defect, most commonly a conotruncal cardiac defect, is seen, information about family history is requested, specifically the presence of velocardialfacial syndrome or a minimal phenotype, such a velopharyngeal insufficiency. An amniocentesis for FISH analysis at locus 22q11.2 would be performed to make a diagnosis [3].
|
| Disease Presentation | DiGeorge Syndrome or 22q11.2 deletion syndrome has many symptoms that can affect almost every region of the body in an affected individual. Features can vary widely among affected individuals, even those from the same family. The most common findings include [9][1]:
Although not as common, it is important to note that 6 cases of females with 22q11.2 deletion syndrome have been reported to have uterovaginal aplasia [10]. |
| Impact on Fertility | Uterovaginal aplasia, or Mayer-Rokitansky-Kuster-Hauser syndrome, has been associated with females with 22q11.2 deletion syndrome and causes the vagina and uterus to be underdeveloped or absent. Although affected women have a normal female karyotype (46,XX) and will develop normal secondary sex characteristics, they will not begin a menstrual period or be able to carry a pregnancy [12].
DiGeorge syndrome patients without immunodeficiency may not be at any increased for infertility. However, in DiGeorge syndrome patients with immunodeficiency due a T-cell deficiency, it can be due to being athymic [11]. Infants with DiGeorge Syndrome can be broken down into 2 categories:
This distinction is important with respect to thymus transplantation because T-cells in an atypical complete DiGeorge anomaly can reject transplants [11]. For individuals with complete DiGeorge anomaly, transplantation of allogeneic postnatal cultured thymus has been successfully studied. Thymus transplants in both typical and atypical complete DiGeorge anomalies resulted in functional immune reconstitution. This was shown by diverse T-cell repertoires and good proliferative function in T-cells [11]. Although the goal to prevent infection and therefore increase life span in DiGeorge syndrome patients is improving, thymus transplantation requires the use of immunosuppressant drugs, to prevent transplant rejection. Unfortunately, immunosuppressant drugs can cause infertility in both men and women. Families of children with DiGeorge syndrome who require transplantation should be informed of fertility preservation options prior to therapy initiation, allowing them to fully consider the options available for their child. |
| Fertility Preservation Options for Women | Oocyte or Embryo Cryopreservation
Ooctyte and embryo cyropreservation are the most effective methods to preserve fertility. Ovarian Tissue Cryopreservation One entire ovary is removed surgically and the outer surface (cortex), which contains the eggs, is frozen in strips for later use. Pieces of the tissue may be thawed and transplanted back into the body. A number of pregnancies have resulted from using this technique. |
| Fertility Preservation Options for Men | Sperm Banking
Sperm cells are collected and frozen for future use. Testicular Tissue Banking Testicular tissue, including cells that produce sperm and sperm itself, is removed and frozen. The procedure has varying risks and side effects. This options may not be appropriate for many patients. |
| Additional Considerations | Once a mutation is present in either a male or female, there is a 50% chance for an affected pregnancy. Although prenatal testing is available, this can pose very difficult decisions for parents whom have an affected pregnancy, as the topic of termination is a controversial one [13]. Genetic counselors are important in this process in order to fully inform families of their options prior to conception.
One alternative is preimplantation genetic diagnosis (PGD). PGD makes it possible for us to analyze embryos for specific genetic disorders [14]. This process involves in vitro fertilization, and can therefore be quite costly and has limited availability. Steps of PGD [14]:
The first case of PGD in a patient with DiGeorge syndrome was reported in 1998 by Iwarsson et al. The proband was a 31-year-old female with no clinical symptoms of DiGeorge Syndrome, but did present with the classic facial features. She received her diagnosis after the birth of a severely affected child, with a 22q11.2 deletion, who died at 4-months of age. None of the three PGD attempts resulted in a pregnancy in this case [15]. In 2009, Shefi et al reported a successful PGD in a 32-year-old woman with DiGeorge syndrome, with typical facial features and velopharyngeal anomaly. She has a 3-year-old daughter who was diagnosed with DiGeorge syndrome shortly after birth. FISH was used to detect 22q11.2 deletions in the embryos, and after three PGD attempts, the family gave birth to a healthy female baby [16]. |
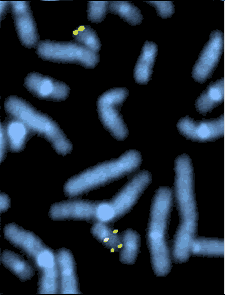 Postnatal diagnosis of DiGeorge syndrome is generally straightforward. A majority of patients with a clinical phenotype of DiGeorge syndrome have a hemizygous deletion of chromosome 22q11.2. The current most accurate method of diagnosis is FISH analysis, however this method can be expensive. MLPA testing has a relatively low cost, and is a suitable screening method for 22q11.2. It includes the determination of the deletion breakpoints in a quantitative method and without the need of pedigree analysis or parental samples. If a patient presents with phenotypic characteristics and no mutation is found through FISH analysis or MLPA, there is a possibility of a point mutation in TBX1, as stated above, which is too small to be picked up on via FISH [8] [6].
Postnatal diagnosis of DiGeorge syndrome is generally straightforward. A majority of patients with a clinical phenotype of DiGeorge syndrome have a hemizygous deletion of chromosome 22q11.2. The current most accurate method of diagnosis is FISH analysis, however this method can be expensive. MLPA testing has a relatively low cost, and is a suitable screening method for 22q11.2. It includes the determination of the deletion breakpoints in a quantitative method and without the need of pedigree analysis or parental samples. If a patient presents with phenotypic characteristics and no mutation is found through FISH analysis or MLPA, there is a possibility of a point mutation in TBX1, as stated above, which is too small to be picked up on via FISH [8] [6].
References
[1] Cancrini, C., Puliafito. P., Digillo M., et al. (2014). Clinical Features and Follow-up in Patients with 22q11.2 Deletion Syndrome. The Journal of Pediatrics, 164(6), 1475-1480.
[2] Grassi, M., Jacob, C., Kulikowski, L., et al. (2014). Congenital Heart Disease as a Warning Sign for the Diagnosis of the 22q11.2 Deletion. Arq Bras Cardiol.
[3] Noel, A., Pelluard, F., Delezoide A., et al. (2014). Fetal Phenotype Associated with the 22q11 Deletion. American Journal of Medical Genetics, 164A, 2724-2731.
[4] Sullivan, K. (2002). Immunologic issues in VCFS/chromosome 22q11.2 deletion syndrome. Progress in Pediatric Cardiology, 15, 103-108.
[5] Shaikh, T., Kurahashi, H., Saitta, S., et al. (2000). Chromosome-22 specific low copy repeats and 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Human Molecular Genetics, 9(4), 489-501.
[6] Yagi, H., Furutani, Y., Hamada, H., et al. (2003). Role of TBX1 in human del22q11.2 syndrome. The Lancet, 362, 1366-1373.
[7] Driscoll, D. (2001). Prenatal diagnosis of the 22q11.2 deletion syndrome. Genetics in Medicine, 3(1), 14-18.
[8] Fernandez L, Lapunzina P, Arjona D, et al. Comparative study of three diagnostic approaches (FISH, STRs and MLPA) in 30 patients with 22q11.2 deletion syndrome. Clin Genet 2005; 68: 373–78.
[9] 22q11.2 deletion syndrome. (2013). U.S. National Library of Medicine. http://ghr.nlm.nih.gov/condition/22q112-deletion-syndrome
[10] Morcel, K., Watrin, T., Pasquier, L., et al. (2011). Utero-vaginal aplasia (Mayer-Rokitansky-Küster-Hauser syndrome) associated with deletions in known DiGeorge or DiGeorge-like loci. Orphanet Journal of Rare Diseases, 6:9.
[11] Markert, M., Devlin, B., Alexieff, M., et al. (2007). Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. American Society of Hematology, 109, 4539-4547.
[12] Mayer-Rokitansky-Kuster-Hauser syndrome. (2010). U.S. National Library of Medicine. http://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrome
[13] McDonald-McGinn, D., Zackai, E. (2008). Genetic counseling for the 22q11.2 deletion. Developmental Disabilites Research Reviews, 14: 69-74.
[14] Pre-Implantation Genetic Diagnosis. (2014) Oregon Health & Science University Center for Women’s Health. http://www.ohsu.edu/xd/health/services/women/services/fertility/fertility-services/fertility-treatment-options/pre-implantation.cfm
[15] Iwarsson, E., Ahrlund-Richter, L., Inzunza, J., et al. (1998). Preimplantion genetic diagnosis of DiGeorge Syndrome. Molecular Human Reproduction, 4(9):871-875.
[16] Shefi, S., Raviv, G., Rienstein, S., et al. (2009). Fish based preimplantation genetic diagnosis to prevent DiGeorge syndrome. J Assist Reprod Genet, 26:411-413.
About the Author
Joanna Lee is a second year Genetic Counseling Master’s student at Northwestern University in the Graduate Program in Genetic Counseling.