
Hurler Syndrome (MPS I)
| Incidence | Hurler syndrome, a Mucopolysaccharidosis type 1 (MPS I) condition, occurs in ~1/100,000 infants born[1]. It is a panethnic condition, affecting individuals all over the world, however there is a higher proportion of infants born with Hurler syndrome in North America and Europe than in Latin America or the Asia Pacific region. This may be due to differences in genetic landscape or the result of regional differences in diagnosis and enrollment of Hurler syndrome patients into the voluntary MPS I registry. The MPS I registry includes 987 MPS I patients worldwide, 60.9% of which have the most severe MPS I disorder, Hurler syndrome[2]. |
| Natural History |  MPS I is a rare autosomal recessive inherited disease caused by a deficiency of alpha-L-iduronidase, an enzyme required for the degradation of the glycosaminoglycans (GAGs) dermatan and heparin sulfate, due to mutations in the IDUA gene. The chronic and progressive accumulation of GAGs in the lysosomes of cells throughout the body leads to multiorgan dysfunction and considerable morbidity. If untreated, patients with Hurler syndrome experience progressive deterioration of the musculoskeletal, cardiorespiratory, and central nervous systems, leading to death before age 10 years[1]. MPS I is a rare autosomal recessive inherited disease caused by a deficiency of alpha-L-iduronidase, an enzyme required for the degradation of the glycosaminoglycans (GAGs) dermatan and heparin sulfate, due to mutations in the IDUA gene. The chronic and progressive accumulation of GAGs in the lysosomes of cells throughout the body leads to multiorgan dysfunction and considerable morbidity. If untreated, patients with Hurler syndrome experience progressive deterioration of the musculoskeletal, cardiorespiratory, and central nervous systems, leading to death before age 10 years[1]. |
| Disease Presentation | MPS I varies significantly with regard to disease presentation and course of disease. This may be due to differences among patients in the severity of the underlying IDUA mutations and consequent residual degree of enzyme activity[3]. Delineation of the different MPS I phenotypes can be challenging[4]; the three MPS I subtypes are:
|
| Effect on Fertility | Since 1980, hematopoietic stem cell transplantation (HSCT) has been used to treat Hurler syndrome.Hurler-Scheie and Scheie syndromes are not traditionally treated with HSCT. Instead, individuals with Hurler-Scheie and Scheie syndromes receive enzyme replacement therapy (ERT) with laronidase (recombinant alpha-L-iduronidase; Aldurazyme) as the primary treatment[2].
HSCT is recommended for patients with Hurler syndrome under the age of 2 years with normal cognitive function (defined as IQ>70)[5]. When successful, it is a one-time procedure that can prolong survival, preserve cognitive function, and reduce morbidity[6]. HSCT reduces Hurler syndrome disease morbidity with substantial clinical benefit, including alleviation of hepatosplenomegaly, upper airway obstruction (including sleep apnea), cardiac symptoms, and coarse facial features. Unfortunately, early diagnosis of Hurler syndrome is limited by several factors, including: rarity of the disease, wide variability in clinical presentation and disease course, and the nonspecific nature of early disease manifestations[5]. The goal of HSCT is to cure the patient from the primary disease and restore complete physical and mental health condition; however, infertility is common after myeloablative HSCT conditioned with total body irradiation (TBI) and high doses of gonadotoxic drugs[7]. Female Fertility
HSCT conditioning often involves TBI and/or gonadotoxic drugs, both of which are associated with a high risk of ovarian failure in females[7,8]. The majority of female patients who are treated with TBI experience gonadal failure. In fact, 10% experience recovery of gonadal function and only 1.3% acheive pregnancy[7]. Vatanen et al. (2013) evaluated long-term ovarian function after allogenic HSCT in childhood and adolescence in 70 prepubertal female tranplant survivors and 22 adult transplant survivors (transplant occuring between 1987-2000; mean age of 9 years; age range 1-19 years). Results showed that 40/70 prepubertal females experienced spontaneous puberty, based on breast development, and 30/70 experience spontaneous menarche. They found that there was a high risk of ovarian failure with conditioning TBI or Bu-based regimes prior to HSCT and intensive anti-leukemia therapy prior to HSCT, including CRT, decreased the possibility of spontaneous menarche. This study was not specific to MPS I. Remerand et al. (2009) reported a female patient with MPS I treated by allogenic HSCT at the age of 4 years (conditioning with busulfan 16mg/kg and cyclophosphamide 100mg/kg) who had 4 successful pregnancies without any reproductive assistance. Therefore, spontaneous pregnancy should not be ruled out as a possibility and contraceptives should be used by women with Hurler syndrome not actvely trying to achieve pregnancy. Male Fertility
TBI and gonadotoxic drugs are typically used to condition patients prior to HSCT, both of which lead to absence of spermatozoa. In fact, TBI is the main cause of azoospermia in patients treated with HSCT. This is due to the fact that spermatogenic stem cells are more sensitive to chemotherapy and radiation than later stage germ cells, preventing the generation of new sperm cells[7]. Rovo et al. (2013) conducted a European Group for Blood and Marrow Transplantation (EBMT) large retrospective analysis of men who underwent HSCT and found that 81% of males receiving TBI presented with azoospermia and only 1% had normal sperm counts in follow-up sperm analysis. Anserini et al. (2002) found that azoospermia is less frequent in patients conditioned with busulphan and cyclophosphamide (50%) and uncommon in those treated with cyclophosphamide alone. Suggesting, perhaps, that busulphan and cyclophosphamiade or cyclophosphamide alone may affect fertility less than traditional HSCT conditioning with TBI and gonadotoxic drugs. |
| Fertility Preservation | Female Fertility Preservation
The risk of premature ovarian failure (POF) and infertility is very high after HSCT, however fertility issues may not become relevant for transplant survivors until a later timepoint. Therefore, fertility preservation discussions should be discussed with all young women of childbearing age and parents of children facing HSCT[12]. The main fertility preservation options available for women include: embryo cryopreservation, oocyte cryopreservation, and ovarian tissue cryopreservation. However, given that children with Hurler syndrome are prepubertal, they cannot undergo the necessary ovarian stimulation needed for embryo and oocyte cryopreservation. Therefore, ovarian tissue cryopreservation and autotransplantation should be considered prior to HSCT in children with Hurler syndrome[9]. Females with Hurler syndrome need to have HSCT prior to 2 years of age, therefore retrieval and preservation must be performed on young children. Given that spontaneous pregnancies can occur after HSCT, there is a risk of decreasing ovarian potential and damaging subsequent gonadal function and fertility with ovarian tissue cryopreservation in young children[9]. Ovarian Tissue Cryopreservation
Male Fertility Preservation Given the risk of azoospermia is very high after HSCT, fertility preservation options should be discussed with all young men of reproductive age and parents of children facing HSCT[7]. Children with Hurler syndrome are prepubertal at the time of HSCT and prepubertal males cannot produce semen for cryopreservation. Testicular tissue cryopreservation is currently the only option for prepubertal males at high risk for infertility interested in preserving future fertility[13]. Ginsberg et al. (2014) consented 57 families to testicular biopsy, and 28 children underwent the procedure with 1 experiencing a post-operative side effect. Parents who faced this fertility preservation option at their child’s diagnosis reported they made an informed decision and carefully weighed the risks and benefits of testicular tissue cryopreservation. Testicular Tissue Cryopreservation Alternatively, males with Hurler syndrome may choose to use donor sperm or consider adoption. |
| Expert Opinion | Tichelli & Rovo (2013):
“Long-term survivors are at risk of late complications interfering with health condition and quality of life after HSCT. Infertility is one among other complications that may occur after transplantation. Today it becomes more evident that patients treated with HSCT are at high risk of permanent infertility. Despite not being a life-threatening complication, infertility is associated with significant psychological distress for the recipient of HSCT and his family. Long-term survivors have a legitimate right of becoming parents of children. Today, there are reasonable options to preserve fertility in patients treated with HSCT. For most late effects that may appear after HSCT, surveillance and preemptive interventions are undertaken during long-term follow-up after transplantation. In contrast, fertility preservation needs to be decided and implemented before starting HSCT. Despite high patient’s interest in issues of fertility, a number of barriers impede clinicians to discuss overtly the risk of infertility and the option for fertility preservation with many patients undergoing HSCT. In addition, providing information and guidance about fertility preservation raises a number of ethical and legal considerations.” |
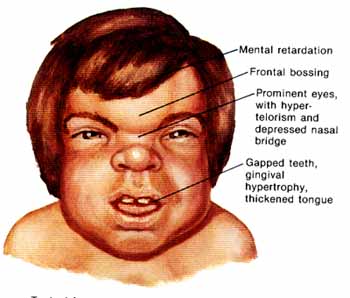 The MPS I registry reports the following findings in individuals with Hurler syndrome
The MPS I registry reports the following findings in individuals with Hurler syndrome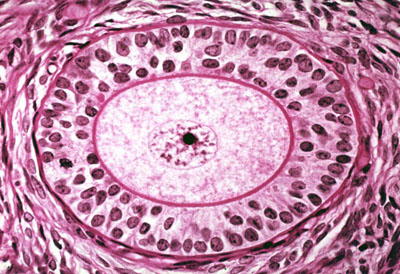
 Alternatively, females with Hurler syndrome may wish to use donor ooctyes or consider adoption.
Alternatively, females with Hurler syndrome may wish to use donor ooctyes or consider adoption.References
[1]Neufeld, E., & Muenzer, J. (2001). The Mucopolysaccharidoses. In C. Scriver, A. Beaudet & W. Sly (Eds.), The Metabolic and Molecular Bases of Inherited Disease (pp. 3421-3452). New York, NY: McGraw Hill.
[2]Beck, M., Arn, P., Giugliani, R., Muenzer, J., Okuyama, T., Taylor, J., & Fallet, S. (2014). The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med.
[3]Terlato, N., & Cox, G. (2003). Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A comprehensive review of the literature. Genet Med, 5, 286-294.
[4]Muenzer, J., Wraith, J., & Clarke, L. (2009). International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics, 123, 19-29.
[5]Peters, C., Balthazor, M., Shapiro, E., King, R., Kollman, C., Hegland, J., . . . Wagner, J. (1996). Outcomes of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood, 87, 4894-4902.Hobbs, J., Hugh-Jones, K., Barrett, A., Byrom, N., Chambers, D., Henry, K., . . . Young, E. (1981). Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet, 318(8249), 709-712.
[6]Hobbs, J., Hugh-Jones, K., Barrett, A., Byrom, N., Chambers, D., Henry, K., . . . Young, E. (1981). Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet, 318(8249), 709-712.
[7]Tichelli, A., & Rovo, A. (2013). Fertility issues following hematopoietic stem cell transplantation. Expert Rev Hematol., 6(4), 375-388.
[8]Vatanen, A., Wilhelmsson, M., Borgstrom, B., Gustafsson, B., Taskinen, M., Saarinen-Pihkala, U., . . . Jahnukainen, K. (2013). Ovarian function after allogenic hematopoietic stem cell transplantation in childhood and adolescence. Eur J Endocrinol, 170(2), 211-218.
[9]Remerand, G., Merlin, E., Froissart, R., Brugnon, F., Kanold, J., Janny, L., & Demeocq, F. (2009). Four successful pregnancies in a patient with mucopolysaccharidosis type I treated by allogeneic bone marrow transplantation. J Inherit Metab Dis, 32(Suppl 1), S111-S113.
[10]Rovo, A., Aljurf, M., Chiodi, S., S, S., Salooja, N., & G, S. (2013). Ongoing graft-versus-host disease is a risk factor for azoospermia after allogeneic hematopoietic stem cell transplantation. A survey of the late effect working party of the European Group for Blood and Marrow Transplantation. Haematologica, 98(3), 339-345.
[11]Anserini, P., Chiodi, S., Spinelli, S., Costa, M., Conte, N., Copello, F., & Bacigalupo, A. (2002). Semen analysis following allogenic bone marrow transplantation. Additional data for evidence-based counselling. Bone Marrow Transplant, 30(7), 447-451.
[12]Jadoul, P., & Donnez, J. (2012). How does bone marrow transplantation affect ovarian function and fertility? Curr. Opin. Obstet. Gynecol., 24(3), 164-171.
[13]Ginsberg, J., Li, Y., Carlson, C., Gracia, C., Hobbie, W., Miller, V., . . . Kolon, T. (2014). Testicular tissue cryopreservation in prepubertal male children: An analysis of parental decision-making. Pediatr Blood Cancer.
About the Author
Allison Goetsch, MS, CGC is a pediatric genetic counselor at Ann and Robert H. Lurie Children’s Hospital of Chicago and a member of the Oncofertility Consortium administrative core team. She completed her graduate thesis with Dr. Teresa K. Woodruff researching oncofertility and hereditary breast and ovarian cancer (HBOC) syndrome. Allison’s primary goals are to increase fertility preservation awareness and education for both health care providers and patients regarding malignant and non-malignant diseases (and/or treatments) which threaten fertility.
This page was last updated February 6th, 2016.